
 Shakir Ali1
Shakir Ali1 2Department of Gastroenterology, Acibadem Dr. Sinasi Can Kadikoy Hospital, Istanbul, Turkiye
3Department of Gastroenterology, Firat University School of Medicine, Elazig, Turkiye

Abstract
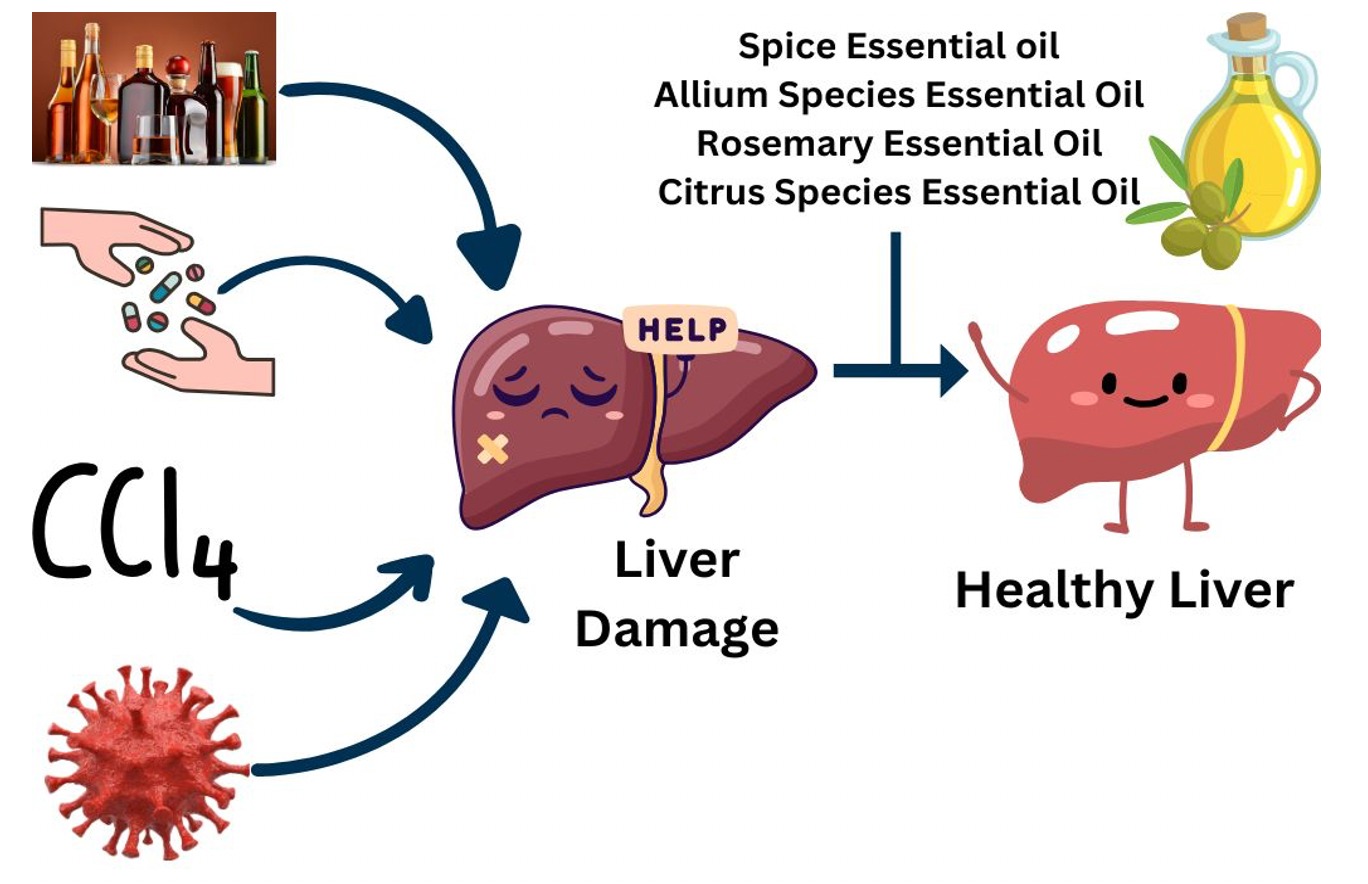
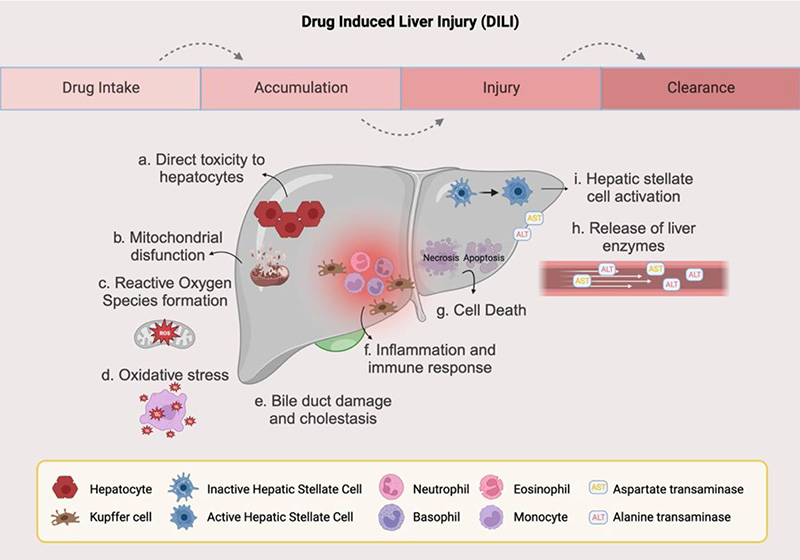
Drug dose efficacy/toxicity depends on a number of factors including genetic and nongenetic factors, a pre-existing disease, and coadministration of other substances and drugs. Cytochrome P450 (CYP) proteins play a crucial role in drug metabolism where they catalyse a number of Phase I oxidation reactions. Concurrently administered drugs and substances, besides the CYP genotype are crucial and can induce/inhibit the CYP activity, thus affecting drug biotransformation and its bioavailability, compromising with drug efficacy, or even causing toxicity due to slow metabolism. Hepatic CYP is particularly important as it metabolizes about ¾ of all drugs. Determining the metabolite/drug ratio (in vivo CYP phenotyping) can be an important tool that can help in drug dose optimization for the drugs metabolized by specific CYPs as the genotype may not always reflect the true enzyme activity. Clinically important CYP isoforms commonly reported in drug oxidation reactions and which mainly include CYP3A4/5, CYP2C19, CYP2C9 and CYP2D6 need to be analysed for their activity in vivo, in at least the cases of unpredictable treatment outcomes. The activity levels of other less commonly reported but no less important CYPs, such as CYP2B6, one of the most polymorphic human CYP involved in the metabolism of artemisinin, bupropion, cyclophosphamide, efavirenz, ketamine and methadone, and reported for its high inter-individuals and within-individual variability may also be determined on a case-to-case basis. This review highlights the variations in CYP activity due to various reasons and the importance of in vivo phenotyping over genotype in ascertaining drug bioavailability and dose optimization, implicating metabolite/drug ratio determination for personalized treatment of especially chronic liver disease patients.








